
Biomolecules such as proteins and protein assemblies perform a strikingly broad range of tasks, from serving as structural elements for keeping the cell in shape to transport material and catalyse chemical reactions. This versatility is largely due to a complex interplay of processes taking place at different length and time scales, making proteins a prominent example of multi-scale soft matter systems.
To investigate in detail the properties that underlie the function of proteins and other biomolecules, it is necessary to take into account their multi-scale nature; this, however, often implies that in the computational study of these systems no simplification of the structure and interactions is possible, as it is done e.g. in coarse-grained models, since the small-scale, chemistry-specific detail plays a crucial role. On the other hand, the size of these molecules might make their computer simulation with a high level of detail unfeasible.
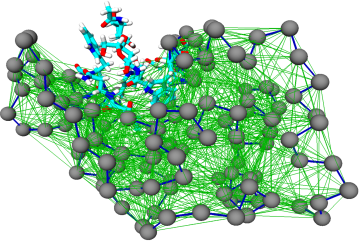
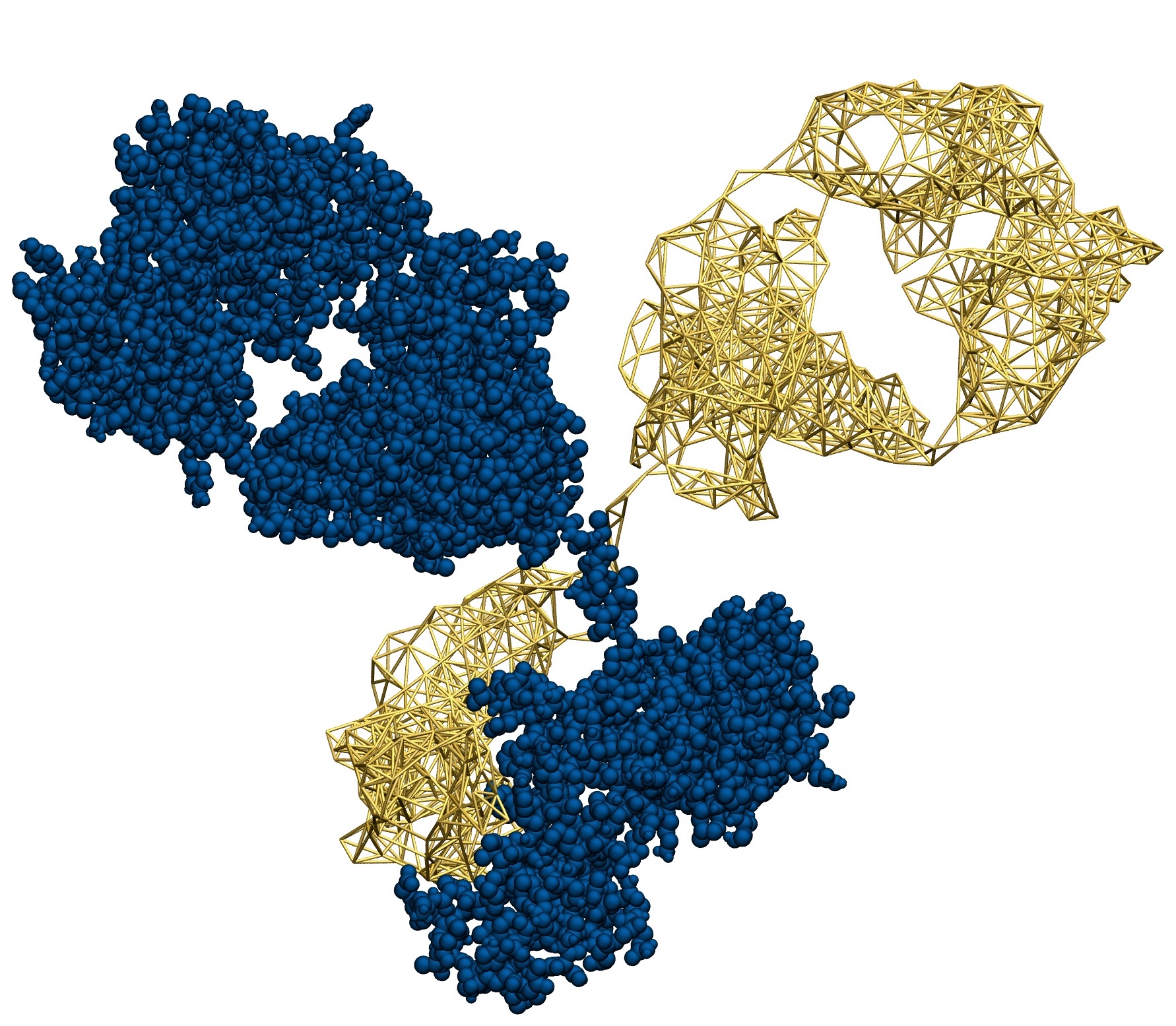
The goal of this research line is to develop algorithmic procedures to construct simplified models of large biomolecules that are at the same time computationally efficient and chemically/functionally accurate. This is done, for example, employing different levels of model resolution in different parts of the system. A theoretically sound and computationally effective solution to this ambitious goal is the objective of the ERC Starting Grant project VARIAMOLS.
Similarly in spirit, it is possible to perform computer simulations of liquids where different models of the same system are employed in separate yet connected regions of the simulation domain. Molecules are free to diffuse across the geometrical boundaries separating regions where a high-resolution and a low-resolution model are used, and change resolution on the fly. These approaches, dubbed adaptive resolution simulations, proved to be particularly effective not only to reduce the computational load of a simulation, rather also and possibly most of all for the quantitative characterisation of the system under examination and the physico/chemical properties of the two models concurrently employed. A research line pursued in our group aims at the development and usage of these methods for the study of biologically relevant liquids such as water and aqueous mixtures.
   |
REPRESENTATIVE BIBLIOGRAPHY
- M. Giulini, M. Rigoli, G. Matiotti, R. Menichetti, T. Tarenzi, R. Fiorentini and Raffaello Potestio, From system modelling to system analysis: the impact of resolution level and resolution distribution in the computer-aided investigation of biomolecules, Front. Mol. Biosci. (2021)
- M. Giulini, R. Menichetti, M. Scott Shell and Raffaello Potestio, An information theory-based approach for optimal model reduction of biomolecules, J. Chem. Theor. Comput. (2020)
- Patrick Diggins, Changjiang Liu, Markus Deserno, and Raffaello Potestio, Optimal coarse-grained site selection in elastic network models of biomolecules, J. Chem. Theor. Comput. (2019)
- Karsten Kreis, Mark E. Tuckerman, Davide Donadio, Kurt Kremer, and Raffaello Potestio, From Classical to Quantum and Back: A Hamiltonian Scheme for Adaptive Multiresolution Classical/Path-Integral Simulations, J. Chem. Theory Comput. 12 (7), 3030-3039 (2016)
- Aoife C. Fogarty, Raffaello Potestio and Kurt Kremer, A multi-resolution model to capture both global fluctuations of an enzyme and molecular recognition in the ligand-binding site, Proteins: Structure, Function, and Bioinformatics, DOI: 10.1002/prot.25173 (2016)
- Karsten Kreis, Aoife C. Fogarty, Kurt Kremer, Raffaello Potestio, Advantages and challenges in coupling an ideal gas to atomistic models in adaptive resolution simulations, Eur. Phys. J. Special Topics, 224, 2289 (2015)
- Sebastian Fritsch, Raffaello Potestio, Davide Donadio, and Kurt Kremer, Nuclear Quantum Effects in Water: A Multiscale Study, J. Chem. Theory Comput. , 10 (2), 816-824 (2014)
- Raffaello Potestio, Christine Peter, and Kurt Kremer, Computer Simulations of Soft Matter: Linking the Scales, Entropy special issue “Molecular Dynamics Simulations” , 16(8), 4199-4245 (2014)
- Raffaello Potestio, S. Fritsch, P. Español, R. Delgado-Buscalioni, K. Kremer, R. Everaers, and D. Donadio,Hamiltonian Adaptive Resolution Simulation for Molecular Liquids, Phys. Rev. Lett. 110 , 108301 (2013)
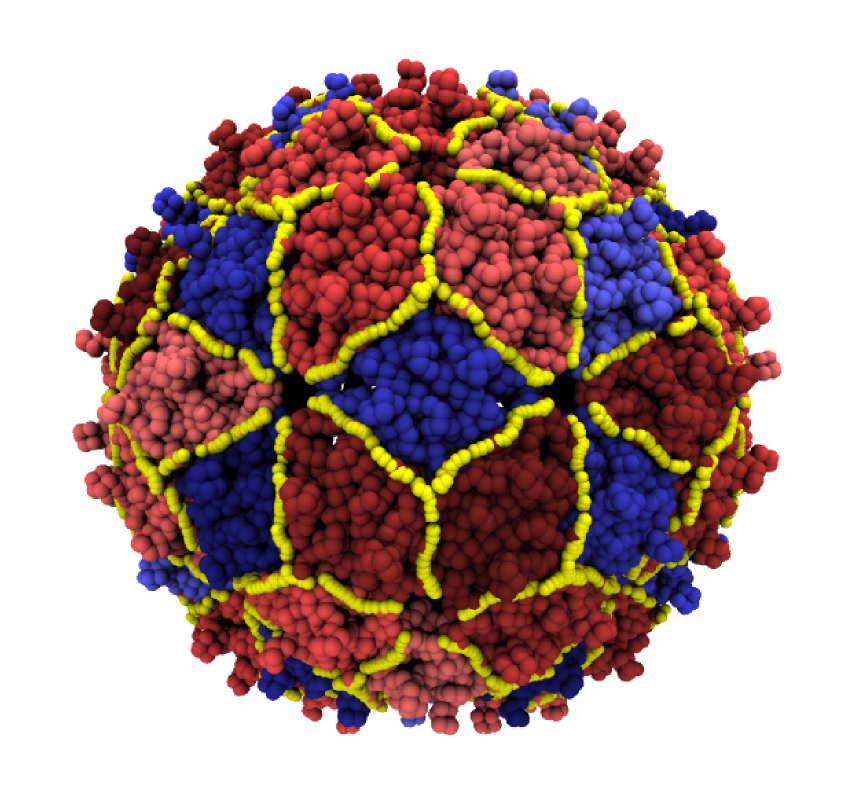
- Guido Polles, Giuliana Indelicato, Raffaello Potestio, Paolo Cermelli, Reidun Twarock, and Cristian Micheletti, Mechanical and Assembly Units of Viral Capsids Identified via Quasi-Rigid Domain Decomposition, PLoS Computational Biology 9(11): e1003331. doi: 10.1371/journal.pcbi.1003331 (2013)
